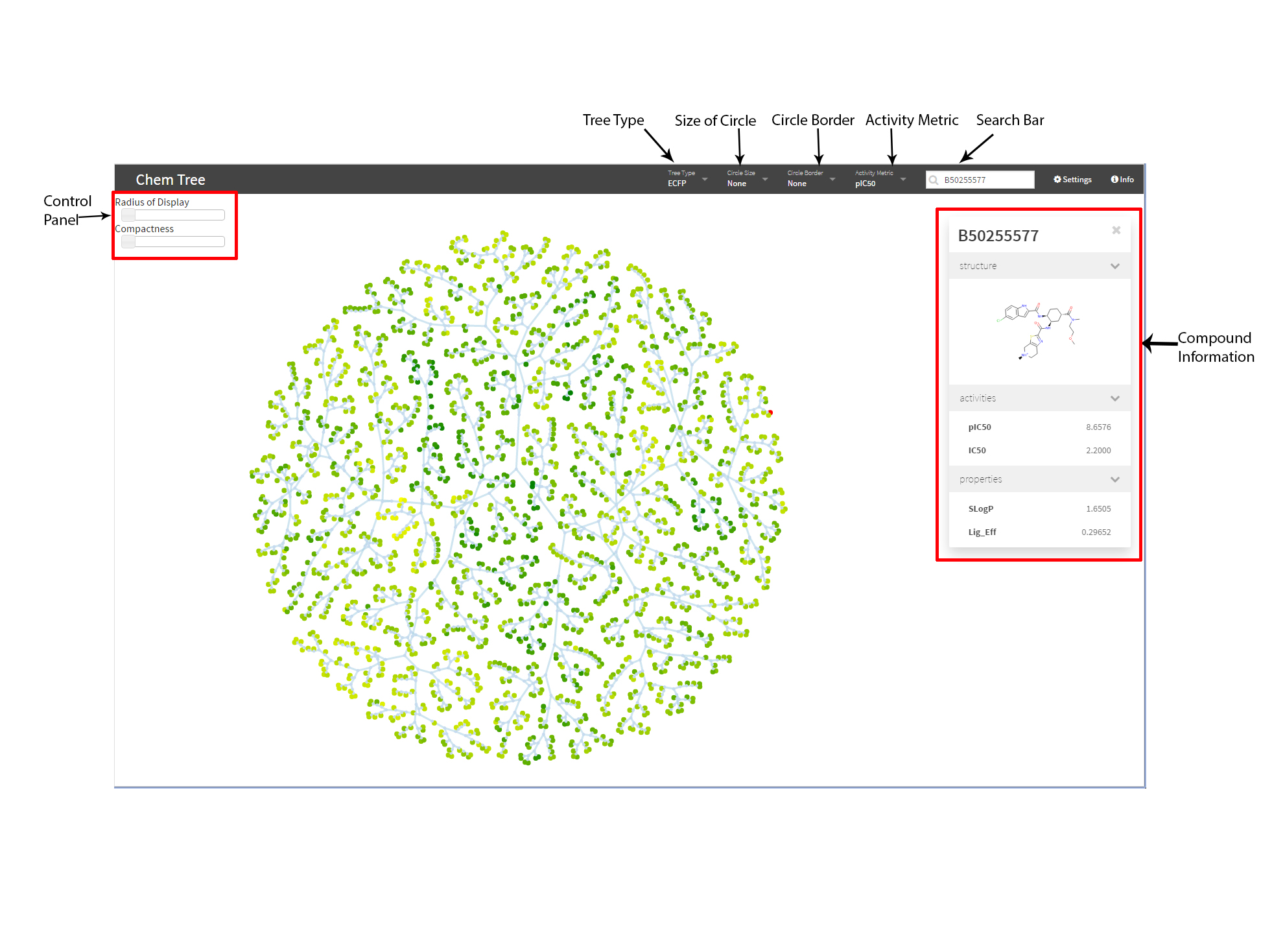
Introduction
ChemTreeMap is an Open Source interactive application for visualizing chemical similarity/diversity in a molecule library.
For detailed documentation, please check ChemTreeMap on readthedocs
Installation
ChemTreeMap contains two parts: frontend (JavaScript) and backend (Python). Users can install ChemTreeMap backend as python package. We support two ways to install ChemTreeMap.
- Docker installation: Run ChemTreeMap in a Docker container with a reproducible environment.
- Source code installation: Several packages are needed to fully install ChemTreeMap. Please check this wiki page for more details.
Docker installation (fastest install)
Docker is a system which can be used to build self contained versions of a Linux operating system running on your machine. When you install and run ChemTreeMap via Docker, it completely isolates the installation from pre-existing packages on your machine.
Please follow the following steps to install ChemTreeMap Docker container. (Superuser privilege may be necessary for the following commands)
See installing Docker for instructions on installing Docker on your machine.
After Docker is installed, pull ChemTreeMap image as follows.
$ docker pull ajing/chemtreemap
Then, launch a Docker container with the binary image as follows.
$ docker run -t -i -p 8000:8000 ajing/chemtreemap /bin/bash
The example code is in /examples folder. Please read and run the examples.py file to see how it works with example input files.
$ cd examples$ python examples.py
Then, open the following URL in your Chrome/Firefox.
http://localhost:8000/dist/#/aff
'aff' may be changed to your input filename without file extension.
If you have your own molecule files, please prepare your input file following the same tab delimited format as example files (e.g. aff.txt, factorxa.txt, etc.). The following command can be used to import files into Docker container.
docker cp foo.txt ajing/chemtreemap:/examples/foo.txt
Please change examples.py (variables input_file, out_file) accordingly and run the examples.py.
Features
- Circle border, size, and color can be mapped to chosen properties in the data.
- Dragging and zooming.
- Searching by ligand ID (PubChem ID or BindingDB ID).
What can you do with ChemTreeMap.
- Comparing multiple molecular libraries
- QSAR studies
- Organize molecule related data
Some tips about using ChemTreeMap.
- We recommend Chrome or Firefox as your browser
- You can use the setting to activate the force directed graph
- After activating force directed graph, the location for each node will be adjusted.
- For more than 3000 nodes, the force directed graph may be slow.
- You can drag the whole tree by dragging the white space, and zoom with middle mouse button.
Examples to use ChemTreeMap.
Examples for the overlap between large datasets (Please use Chrome or Firefox for large datasets)
Examples for SAR analysis
- A simple example for 107 compounds targeting Chk1 kinase from CSAR. You may not find the external links for BindingDB or PubChem, because the data source doesn't have those external information.
- factor Xa with 2161 molecules.
- cdk2 with 1923 molecules.
- MAP P38 with 5139 molecules.
- cytochrome P450 with 6837 molecules. The input file is about 3.2MB, which may take a few seconds to load into browser.
Who am I?
I am @ajing, a Bioinformatics PhD student from University of Michigan. I am working under the supervision of Dr. Heather Carlson's lab. If you have any question about the project or find any bug, please check out the Wiki first, or contact ajing.
Technologies
- jQuery for client side effects.
- AngularJS for organizing the client side.
- D3 for data visualization.
- I have used some codes from synergy-maps
Citation
If you use our tool, please cite:
ChemTreeMap: An Interactive Map of Biochemical Similarity in Molecular Datasets. submitted to Bioinformatics